Zespół ten wywołany delecją materiału genetycznego z regionu q11.23 chromosomu 7. Usunięty region obejmuje więcej niż 25 genów. Szczególną cechą wyglądu osób dotkniętych syndromem Williamsa jest charakterystyczna dysmorfia twarzy – tzw. "twarz elfa". Osoby dotknięte zespołem Williamsa mogą mieć intelekt mieszczący się w szerokich granicach IQ od 20 do 106. Ludzie dotknięci zespołem Williamsa charakteryzują się tzw. "mową koktajlową" – zdolności językowe są najczęściej obniżone w zakresie semantyki, morfologii, fonologii, ale nie słownictwa. Jest to łatwo zauważalne dla otoczenia, z powodu bogatego słownictwa używanemu przez dzieci dotkniętych tym zespołem. Prawdopodobnie wynika to z połączenia dobrej pamięci słuchowej i trudności z przetwarzaniem języka, co prowadzi do zapamiętywania języka w postaci gotowych zwrotów.Pacjenci z zespołem Williamsa mają charakterystyczną dysmorfię twarzy, określaną jako "twarz elfa", na którą składają się charakterystyczne:
-małżowiny uszne
-szerokie czoło
-długa rynienka podnosowa
-grube wargi
-pogłębiona nasada nosa
-niebieskie lub zielone tęczówki





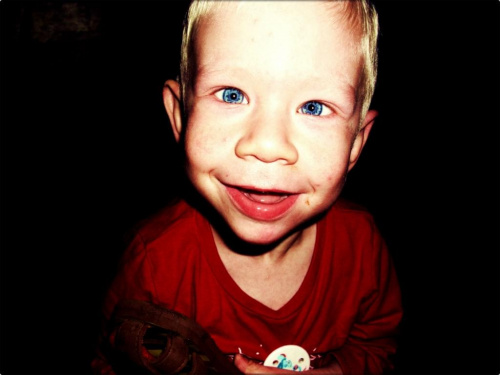
Często, lecz nie zawsze występują u nich :
-wady wrodzone serca (np. nadzastawkowe zwężenie aorty)
-niedoczynność tarczycy
-niepełnosprawność intelektualna lub inteligencja w dolnych granicach normy
-niezwykle życzliwe usposobienie
-zamiłowanie do muzyki.
Zdarza się również nadwrażliwość na dźwięki, która w okresie dorosłości może prowadzić do głuchoty. U osób z zespołem Williamsa często stwierdza się słuch absolutny. W okresie noworodkowym może wystąpić hiperkalcemia. Pacjenci z zespołem Williamsa mają powiększone ciało migdałowate (odpowiadające m.in. za kontrolę strachu) i w związku z tym wykazują zwiększoną ufność wobec nieznanych wcześniej osób. Osoby z zespołem Williamsa mogą też przejawiać inne zaburzenia zachowania i zaburzenia psychiczne, takie jak hiperaktywność, mania i skłonność do nieuzasadnionych lęków przed niektórymi sytuacjami.
Hemofilia
Jest to choroba recesywna powodująca brak krzepliwości krwi, zwana też krwawiączką albo chorobą królów. Wadliwy gen znajduje się na chromosomie X, dlatego też u mężczyzn nie ma możliwości zdominowania go przez gen z homologicznego chromosomu (mają genotyp XY). Z tego powodu wystarczy, by jedno z rodziców przekazało ten gen dziecku, aby zachorował mężczyzna, lecz w przypadku kobiet oboje rodzice muszą posiadać wadliwy gen.
Objawy:
-Krwawienia wewnętrzne:
a)krwawienia podskórne (tzw. siniaki)
b)krwawienia do stawów szczególnie kolanowych, skokowych i łokciowych,
c)krwawienia do mięśni np mięśnia biodrowo-lędźwiowego, łydki,
krwiomocz
-Krwawienia zewnętrzne (rzadziej):
a)krwawienia z błony śluzowej jamy ustnej i nosa
-Krwawienia które mogą zagrażać życiu:
a)krwawienie pozaotrzewnowe,
b)krwawienie do ośrodkowego układu nerwowego,
c)krwawienie w okolicach szyi
Z hemofilią wiąże się wiele całkowicie błędnych poglądów:
-Nieprawdą jest, że "chory na hemofilię umiera bardzo młodo, bowiem każde skaleczenie jest śmiertelnie niebezpieczne". Drobne skaleczenia na ogół nie trwają dłużej niż u innych osób. Głębsze wymagają uciśnięcia i to skutecznie pomaga powstrzymywać krwawienia (w przypadku poważniejszych ran niezbędne jest jednak leczenie farmakologiczne czynnikiem VIII).
-Również błędne jest stwierdzenie, że "chorzy na hemofilię są skazani na życie w domu". Z hemofilią można sobie doskonale radzić, zwłaszcza, jeśli podejmie się leczenie odpowiednio wcześnie. Dzięki nowoczesnemu leczeniu chorzy na hemofilię mogą prowadzić normalne i produktywne życie.
-Błędny także jest pogląd, że "dzieci hemofilika i osoby zdrowej będą na pewno chore". Jeśli mężczyzna jest chory, a kobieta zdrowa i nie jest nosicielką, to wszyscy chłopcy będą zdrowi, zaś dziewczynki będą nosicielkami (dziedziczą chromosom X z uszkodzonym genem po ojcu). Zaś jeśli ojciec jest zdrowy, a matka jest nosicielką, to prawdopodobieństwo, że chłopiec będzie chory, a dziewczynka będzie nosicielką wynosi 1/2 (50%) – dotyczy odmian typu A i B.
 |
| Krzyżówka przenoszenia choroby |
Objawy:
-Krwawienia wewnętrzne:
a)krwawienia podskórne (tzw. siniaki)
b)krwawienia do stawów szczególnie kolanowych, skokowych i łokciowych,
c)krwawienia do mięśni np mięśnia biodrowo-lędźwiowego, łydki,
krwiomocz
-Krwawienia zewnętrzne (rzadziej):
a)krwawienia z błony śluzowej jamy ustnej i nosa
-Krwawienia które mogą zagrażać życiu:
a)krwawienie pozaotrzewnowe,
b)krwawienie do ośrodkowego układu nerwowego,
c)krwawienie w okolicach szyi
Z hemofilią wiąże się wiele całkowicie błędnych poglądów:
-Nieprawdą jest, że "chory na hemofilię umiera bardzo młodo, bowiem każde skaleczenie jest śmiertelnie niebezpieczne". Drobne skaleczenia na ogół nie trwają dłużej niż u innych osób. Głębsze wymagają uciśnięcia i to skutecznie pomaga powstrzymywać krwawienia (w przypadku poważniejszych ran niezbędne jest jednak leczenie farmakologiczne czynnikiem VIII).
-Również błędne jest stwierdzenie, że "chorzy na hemofilię są skazani na życie w domu". Z hemofilią można sobie doskonale radzić, zwłaszcza, jeśli podejmie się leczenie odpowiednio wcześnie. Dzięki nowoczesnemu leczeniu chorzy na hemofilię mogą prowadzić normalne i produktywne życie.
-Błędny także jest pogląd, że "dzieci hemofilika i osoby zdrowej będą na pewno chore". Jeśli mężczyzna jest chory, a kobieta zdrowa i nie jest nosicielką, to wszyscy chłopcy będą zdrowi, zaś dziewczynki będą nosicielkami (dziedziczą chromosom X z uszkodzonym genem po ojcu). Zaś jeśli ojciec jest zdrowy, a matka jest nosicielką, to prawdopodobieństwo, że chłopiec będzie chory, a dziewczynka będzie nosicielką wynosi 1/2 (50%) – dotyczy odmian typu A i B.
Brak komentarzy:
Prześlij komentarz