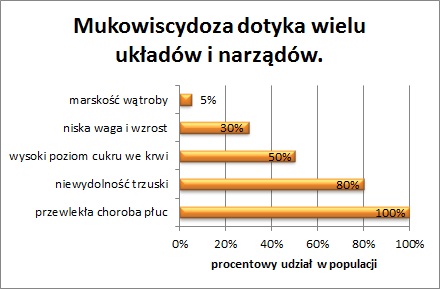
Schorzenie to najczęściej powoduje zmiany w:
a)układzie oddechowym - nawracające zakażenia, które prowadzą do uszkodzenia płuc i niewydolności oddechowej;
b)przewodzie pokarmowym - przewlekły stan zapalny trzustki, prowadzi do uszkodzenia tego narządu i jego niewydolności, a niekiedy także wtórnej cukrzycy.
 |
| Przewlekłe zapalenie płuc ze zniszczeniem miąższu płucnego u osoby z mukowiscydozą |
U ponad 90% chorych występują objawy ze strony układu oddechowego, takie jak:
-przewlekły i napadowy kaszel,
-nawracające i przewlekłe zapalenia płuc,
-zapalenie oskrzelików,
-obturacyjne zapalenia oskrzeli,
-krwioplucie,
-przewlekłe zakażenie Pseudomonas aeruginosa i (lub) Staphylococcus aureus,
-zmiany w płucach widoczne w rtg: nawracająca niedodma, rozdęcie i rozstrzenie oskrzeli,
-polipy nosa,
-przewlekłe zapalenie zatok przynosowych.

Objawy ze strony przewodu pokarmowego występują u około 75% chorych:
-występują obfite, nieuformowane, cuchnące, tłuszczowe stolce od wczesnego dzieciństwa;
-powiększenie objętości brzucha, niekiedy wypadanie odbytnicy;
-niedrożność smółkowa (meconium ileus) jelit w okresie noworodkowym, spowodowana czopem gęstej smółki zatykającym jelito grube i jej ekwiwalenty:
a)zespół korka smółkowego
b)nawracające epizody bólu brzucha z objawami niedrożności przepuszczającej określane jako dystalna niedrożność jelit (DIOS, distal intestinal obstruction syndrome);
-może wystąpić (4-5% przypadków) wtórna marskość żółciowa wątroby z powodu niedrożności kanalików żółciowych;
-kamica żółciowa;
-zaczopowanie przewodów ślinianek gęstą wydzieliną śluzową;
-skręt jelita w okresie płodowym;
-gęsty i lepki śluz blokuje przewody trzustkowe, przyjmowane pokarmy nie są odpowiednio trawione (stąd występowanie stolców tłuszczowych – steatorrhoea) doprowadzając do objawów zespołu złego wchłaniania;
-nawracające zapalenia trzustki.
Mukowiscydoza może powodować bezpłodność. U kobiet ze względu na wzrost gęstości śluzu szyjkowego, co implikuje trudności w przechodzeniu plemników w kierunku komórki jajowej; u mężczyzn związana z oligospermią lub azoospermią spowodowaną niedrożnością i aplazją nasieniowodów, określanym jako CBAVD (congenital bilateral aplasia of vas deferens).
W wyniku zmian płucnych i zwiększonego oporu krążenia płucnego może dojść do powstania serca płucnego ,oraz:
-palce pałeczkowate,
-nawracający obrzęk ślinianek przyusznych.
-osteoporoza